Prácticamente toda la medicina se dedica a prevenir la muerte celular, en enfermedades como el infarto de miocardio, los accidentes cerebrovasculares, los traumatismos y el shock, oa provocarla, como en el caso de las enfermedades infecciosas y el cáncer. Por lo tanto, es esencial comprender la naturaleza y los mecanismos involucrados. La muerte celular se ha clasificado como “accidental”, es decir, causada por agentes tóxicos, isquemia, etc., o “programada”, como ocurre durante el desarrollo embriológico, incluida la formación de dedos y la reabsorción de la cola del renacuajo.
La lesión celular y la muerte celular son, por lo tanto, importantes tanto en fisiología como en fisiopatología. La muerte celular fisiológica es extremadamente importante durante la embriogénesis y el desarrollo embrionario. El estudio de la muerte celular durante el desarrollo ha dado lugar a información nueva e importante sobre la genética molecular involucrada, especialmente a través del estudio del desarrollo en animales invertebrados. En estos animales, se ha estudiado cuidadosamente la ubicación precisa y el significado de las células que están destinadas a sufrir muerte celular y, con el uso de técnicas clásicas de mutagénesis, ahora se han identificado varios genes involucrados. En los órganos adultos, el equilibrio entre la muerte celular y la proliferación celular controla el tamaño del órgano. En algunos órganos, como la piel y el intestino, hay un recambio continuo de células. En la piel, por ejemplo, las células se diferencian a medida que alcanzan la superficie y finalmente experimentan una diferenciación terminal y muerte celular a medida que avanza la queratinización con la formación de envolturas entrecruzadas.
Muchas clases de productos químicos tóxicos son capaces de inducir una lesión celular aguda seguida de la muerte. Estos incluyen la anoxia y la isquemia y sus análogos químicos como el cianuro de potasio; carcinógenos químicos, que forman electrófilos que se unen covalentemente a proteínas en ácidos nucleicos; productos químicos oxidantes, que dan como resultado la formación de radicales libres y lesiones oxidantes; activación del complemento; y una variedad de ionóforos de calcio. La muerte celular también es un componente importante de la carcinogénesis química; muchos carcinógenos químicos completos, en dosis cancerígenas, producen necrosis aguda e inflamación seguidas de regeneración y preneoplasia.
Definiciones
Daño celular
La lesión celular se define como un evento o estímulo, como una sustancia química tóxica, que perturba la homeostasis normal de la célula, provocando así una serie de eventos (figura 1). Los objetivos principales de la lesión letal ilustrados son la inhibición de la síntesis de ATP, la alteración de la integridad de la membrana plasmática o la retirada de factores de crecimiento esenciales.
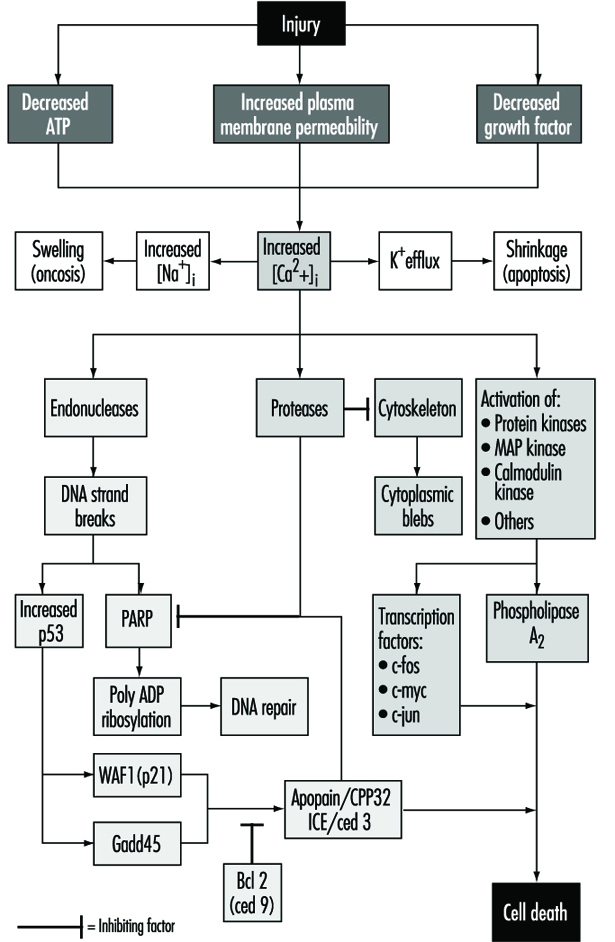
Las lesiones letales provocan la muerte de una célula después de un período de tiempo variable, según la temperatura, el tipo de célula y el estímulo; o pueden ser subletales o crónicos, es decir, la lesión da como resultado un estado homeostático alterado que, aunque anormal, no da como resultado la muerte celular (Trump y Arstila 1971; Trump y Berezesky 1992; Trump y Berezesky 1995; Trump, Berezesky y Osornio-Vargas 1981). En el caso de una lesión letal, existe una fase previa al momento de la muerte celular
durante este tiempo, la célula se recuperará; sin embargo, después de un punto particular en el tiempo (el "punto de no retorno" o punto de muerte celular), la eliminación de la lesión no da como resultado la recuperación, sino que la célula sufre degradación e hidrólisis, alcanzando finalmente el equilibrio físico-químico con el resto. ambiente. Esta es la fase conocida como necrosis. Durante la fase preletal ocurren varios tipos principales de cambios, según la célula y el tipo de lesión. Estos se conocen como apoptosis y oncosis.
La apoptosis
Apoptosis se deriva de las palabras griegas apo, que significa lejos de, y ptosis, que significa caer. El termino cayendo lejos de se deriva del hecho de que, durante este tipo de cambio preletal, las células se encogen y experimentan una marcada ampolla en la periferia. Luego, las ampollas se desprenden y se alejan flotando. La apoptosis ocurre en una variedad de tipos de células después de varios tipos de lesiones tóxicas (Wyllie, Kerr y Currie 1980). Es especialmente prominente en los linfocitos, donde es el mecanismo predominante para el recambio de clones de linfocitos. Los fragmentos resultantes dan como resultado los cuerpos basófilos que se ven dentro de los macrófagos en los ganglios linfáticos. En otros órganos, la apoptosis ocurre típicamente en células individuales que se eliminan rápidamente antes y después de la muerte por fagocitosis de los fragmentos por células parenquimatosas adyacentes o por macrófagos. La apoptosis que ocurre en células individuales con la fagocitosis subsiguiente típicamente no da como resultado inflamación. Antes de la muerte, las células apoptóticas muestran un citosol muy denso con mitocondrias normales o condensadas. El retículo endoplásmico (ER) es normal o solo ligeramente dilatado. La cromatina nuclear está marcadamente agrupada a lo largo de la envoltura nuclear y alrededor del nucléolo. El contorno nuclear también es irregular y se produce fragmentación nuclear. La condensación de la cromatina está asociada con la fragmentación del ADN que, en muchos casos, ocurre entre los nucleosomas, dando una apariencia característica de escalera en la electroforesis.
En apoptosis, aumento de [Ca2+]i puede estimular K+ flujo de salida que resulta en el encogimiento de las células, lo que probablemente requiere ATP. Por lo tanto, es más probable que las lesiones que inhiben totalmente la síntesis de ATP den lugar a la apoptosis. Un aumento sostenido de [Ca2+]i tiene una serie de efectos nocivos que incluyen la activación de proteasas, endonucleasas y fosfolipasas. La activación de la endonucleasa da como resultado roturas de cadena simple y doble de ADN que, a su vez, estimulan niveles elevados de p53 y en la ribosilación de poli-ADP, y de proteínas nucleares que son esenciales en la reparación del ADN. La activación de las proteasas modifica una serie de sustratos, incluida la actina y las proteínas relacionadas, lo que conduce a la formación de vesículas. Otro sustrato importante es la poli(ADP-ribosa) polimerasa (PARP), que inhibe la reparación del ADN. Aumento de [Ca2+]i también se asocia con la activación de varias proteínas quinasas, como MAP quinasa, calmodulina quinasa y otras. Tales quinasas están involucradas en la activación de factores de transcripción que inician la transcripción de genes tempranos inmediatos, por ejemplo, c-fos, c-jun y c-myc, y en la activación de la fosfolipasa A.2 lo que da como resultado la permeabilización de la membrana plasmática y de las membranas intracelulares, como la membrana interna de las mitocondrias.
oncosis
Oncosis, derivado de la palabra griega Es s, hincharse, se llama así porque en este tipo de cambio preletal la célula comienza a hincharse casi inmediatamente después de la lesión (Majno y Joris 1995). La razón de la hinchazón es un aumento de cationes en el agua dentro de la célula. El principal catión responsable es el sodio, que normalmente se regula para mantener el volumen celular. Sin embargo, en ausencia de ATP o si se inhibe la Na-ATPasa del plasmalema, se pierde el control del volumen debido a la proteína intracelular y al continuo aumento del sodio en el agua. Entre los primeros eventos en la oncosis son, por lo tanto, el aumento de [Na+]i lo que conduce a la inflamación celular y al aumento de [Ca2+]i como resultado de la entrada desde el espacio extracelular o la liberación de los depósitos intracelulares. Esto da como resultado la hinchazón del citosol, la hinchazón del retículo endoplásmico y el aparato de Golgi, y la formación de vesículas acuosas alrededor de la superficie celular. Las mitocondrias inicialmente experimentan condensación, pero luego también muestran una gran hinchazón debido al daño a la membrana mitocondrial interna. En este tipo de cambio preletal, la cromatina se condensa y finalmente se degrada; sin embargo, no se observa el patrón en escalera característico de la apoptosis.
Necrosis
La necrosis se refiere a la serie de cambios que ocurren después de la muerte celular cuando la célula se convierte en desechos que normalmente se eliminan mediante la respuesta inflamatoria. Se pueden distinguir dos tipos: necrosis oncótica y necrosis apoptótica. La necrosis oncótica generalmente ocurre en zonas grandes, por ejemplo, en un infarto de miocardio o regionalmente en un órgano después de una toxicidad química, como el túbulo proximal renal después de la administración de HgCl.2. Están afectadas amplias zonas de un órgano y las células necróticas provocan rápidamente una reacción inflamatoria, primero aguda y luego crónica. En el caso de que el organismo sobreviva, en muchos órganos a la necrosis le sigue la eliminación de las células muertas y la regeneración, por ejemplo, en el hígado o el riñón después de la toxicidad química. Por el contrario, la necrosis apoptótica se produce normalmente en una sola célula y los restos necróticos se forman dentro de los fagocitos de los macrófagos o de las células parenquimatosas adyacentes. Las primeras características de las células necróticas incluyen interrupciones en la continuidad de la membrana plasmática y la aparición de densidades floculantes, que representan proteínas desnaturalizadas dentro de la matriz mitocondrial. En algunas formas de lesión que inicialmente no interfieren con la acumulación de calcio mitocondrial, se pueden observar depósitos de fosfato de calcio dentro de las mitocondrias. Otros sistemas de membrana se fragmentan de manera similar, como el RE, los lisosomas y el aparato de Golgi. Finalmente, la cromatina nuclear sufre lisis, como resultado del ataque de las hidrolasas lisosomales. Después de la muerte celular, las hidrolasas lisosomales desempeñan un papel importante en la eliminación de desechos con catepsinas, nucleolasas y lipasas, ya que estas tienen un pH ácido óptimo y pueden sobrevivir al bajo pH de las células necróticas mientras que otras enzimas celulares se desnaturalizan e inactivan.
Mecanismos
Estímulo inicial
En el caso de lesiones letales, las interacciones iniciales más comunes que dan lugar a lesiones que conducen a la muerte celular son la interferencia con el metabolismo energético, como anoxia, isquemia o inhibidores de la respiración, y la glucólisis, como cianuro de potasio, monóxido de carbono, yodoacetato y pronto. Como se mencionó anteriormente, las altas dosis de compuestos que inhiben el metabolismo energético suelen provocar oncosis. El otro tipo común de lesión inicial que resulta en muerte celular aguda es la modificación de la función de la membrana plasmática (Trump y Arstila 1971; Trump, Berezesky y Osornio-Vargas 1981). Esto puede ser daño directo y permeabilización, como en el caso de un trauma o activación del complejo C5b-C9 del complemento, daño mecánico a la membrana celular o inhibición del sodio-potasio (Na+-K+) bombear con glucósidos como la ouabaína. Ionóforos de calcio como la ionomicina o A23187, que transportan rápidamente [Ca2+] por el gradiente en la célula, también causan lesiones letales agudas. En algunos casos, el patrón en el cambio preletal es la apoptosis; en otros, es oncosis.
Vías de señalización
Con muchos tipos de lesiones, la respiración mitocondrial y la fosforilación oxidativa se ven afectadas rápidamente. En algunas células, esto estimula la glucólisis anaeróbica, que es capaz de mantener ATP, pero con muchas lesiones esto se inhibe. La falta de ATP da como resultado la falta de activación de varios procesos homeostáticos importantes, en particular, el control de la homeostasis de iones intracelulares (Trump y Berezesky 1992; Trump, Berezesky y Osornio-Vargas 1981). Esto resulta en rápidos aumentos de [Ca2+]i, y aumentó [Na+] y [Cl-] da como resultado la inflamación de las células. Aumentos en [Ca2+]i dan como resultado la activación de una serie de otros mecanismos de señalización que se analizan a continuación, incluida una serie de quinasas, que pueden dar como resultado un aumento inmediato de la transcripción temprana de genes. Aumento de [Ca2+]i también modifica la función del citoesqueleto, lo que en parte da como resultado la formación de vesículas y la activación de endonucleasas, proteasas y fosfolipasas. Estos parecen desencadenar muchos de los efectos importantes discutidos anteriormente, como el daño de la membrana a través de la activación de la proteasa y la lipasa, la degradación directa del ADN por la activación de la endonucleasa y la activación de quinasas como MAP quinasa y calmodulina quinasa, que actúan como factores de transcripción.
A través de un extenso trabajo sobre el desarrollo en los invertebrados C. elegans y Drosophila, además de células humanas y animales, se han identificado una serie de genes pro-muerte. Se ha descubierto que algunos de estos genes de invertebrados tienen homólogos de mamíferos. Por ejemplo, el gen ced-3, que es esencial para la muerte celular programada en c. elegans, tiene actividad de proteasa y una fuerte homología con la enzima convertidora de interleucina de mamíferos (ICE). Recientemente se ha identificado un gen estrechamente relacionado denominado apopaína o prICE con una homología aún más estrecha (Nicholson et al. 1995). En Drosophila, el gen segador parece estar involucrado en una señal que conduce a la muerte celular programada. Otros genes favorables a la muerte incluyen la proteína de membrana Fas y el importante gen supresor de tumores, p53, que está ampliamente conservado. p53 se induce a nivel de proteína después del daño en el ADN y cuando se fosforila actúa como un factor de transcripción para otros genes como gadd45 y waf-1, que están involucrados en la señalización de muerte celular. Otros genes tempranos inmediatos como c-fos, c-jun y c-myc también parecen estar involucrados en algunos sistemas.
Al mismo tiempo, existen genes anti-muerte que parecen contrarrestar los genes pro-muerte. El primero de ellos en ser identificado fue el ced-9 de C. elegans, que es homólogo a bcl-2 en humanos. Estos genes actúan de una manera aún desconocida para prevenir la muerte celular por toxinas genéticas o químicas. Cierta evidencia reciente indica que bcl-2 puede actuar como un antioxidante. Actualmente, se están realizando muchos esfuerzos para desarrollar una comprensión de los genes involucrados y desarrollar formas de activar o inhibir estos genes, según la situación.