Durante mucho tiempo se ha reconocido que la respuesta de cada persona a los productos químicos ambientales es diferente. La reciente explosión en biología molecular y genética ha traído una comprensión más clara sobre la base molecular de tal variabilidad. Los principales determinantes de la respuesta individual a los productos químicos incluyen diferencias importantes entre más de una docena de superfamilias de enzimas, denominadas colectivamente xenobiótico- (ajena al cuerpo) o metabolizador de drogas enzimas Aunque el papel de estas enzimas se ha considerado clásicamente como desintoxicante, estas mismas enzimas también convierten una serie de compuestos inertes en intermediarios altamente tóxicos. Recientemente, se han identificado muchas diferencias sutiles y grandes en los genes que codifican estas enzimas, que se ha demostrado que dan como resultado marcadas variaciones en la actividad enzimática. Ahora está claro que cada individuo posee un complemento distinto de actividades enzimáticas metabolizadoras de xenobióticos; esta diversidad podría considerarse como una "huella digital metabólica". Es la interacción compleja de estas muchas superfamilias de enzimas diferentes lo que determina en última instancia no solo el destino y el potencial de toxicidad de una sustancia química en un individuo determinado, sino también la evaluación de la exposición. En este artículo, hemos optado por utilizar la superfamilia de enzimas del citocromo P450 para ilustrar el notable progreso realizado en la comprensión de la respuesta individual a los productos químicos. El desarrollo de pruebas relativamente simples basadas en el ADN diseñadas para identificar alteraciones genéticas específicas en estas enzimas ahora proporciona predicciones más precisas de la respuesta individual a la exposición química. Esperamos que el resultado sea una toxicología preventiva. En otras palabras, cada individuo podría aprender acerca de aquellos químicos a los que él o ella es particularmente sensible, evitando así la toxicidad o el cáncer que antes eran impredecibles.
Aunque generalmente no se aprecia, los seres humanos estamos expuestos diariamente a un aluvión de innumerables y diversos productos químicos. Muchos de estos químicos son altamente tóxicos y se derivan de una amplia variedad de fuentes ambientales y dietéticas. La relación entre dichas exposiciones y la salud humana ha sido y continúa siendo un foco importante de los esfuerzos de investigación biomédica en todo el mundo.
¿Cuáles son algunos ejemplos de este bombardeo químico? Se han aislado y caracterizado más de 400 sustancias químicas del vino tinto. Se estima que un cigarrillo encendido produce al menos 1,000 sustancias químicas. Hay innumerables productos químicos en los cosméticos y jabones perfumados. Otra fuente importante de exposición química es la agricultura: solo en los Estados Unidos, las tierras de cultivo reciben más de 75,000 XNUMX productos químicos cada año en forma de pesticidas, herbicidas y agentes fertilizantes; después de que las plantas y los animales de pastoreo los absorban, así como los peces en las vías fluviales cercanas, los humanos (al final de la cadena alimentaria) ingieren estos químicos. Otras dos fuentes de grandes concentraciones de sustancias químicas que ingresan al cuerpo incluyen (a) las drogas que se toman de forma crónica y (b) la exposición a sustancias peligrosas en el lugar de trabajo durante toda la vida laboral.
Ahora está bien establecido que la exposición química puede afectar negativamente muchos aspectos de la salud humana, causando enfermedades crónicas y el desarrollo de muchos tipos de cáncer. En la última década más o menos, la base molecular de muchas de estas relaciones ha comenzado a descifrarse. Además, ha surgido la comprensión de que los seres humanos difieren notablemente en su susceptibilidad a los efectos nocivos de la exposición química.
Los esfuerzos actuales para predecir la respuesta humana a la exposición química combinan dos enfoques fundamentales (figura 1): monitorear el alcance de la exposición humana a través de marcadores biológicos (biomarcadores) y predecir la respuesta probable de un individuo a un nivel dado de exposición. Aunque ambos enfoques son extremadamente importantes, se debe enfatizar que los dos son claramente diferentes entre sí. Este artículo se centrará en la factores genéticos susceptibilidad individual subyacente a cualquier exposición química en particular. Este campo de investigación se denomina ampliamente ecogenéticao farmacogenética (ver Kalow 1962 y 1992). Muchos de los avances recientes en la determinación de la susceptibilidad individual a la toxicidad química han evolucionado a partir de una mayor apreciación de los procesos por los cuales los humanos y otros mamíferos desintoxican las sustancias químicas y la notable complejidad de los sistemas enzimáticos involucrados.
Figura 1. Las interrelaciones entre la evaluación de la exposición, las diferencias étnicas, la edad, la dieta, la nutrición y la evaluación de la susceptibilidad genética, todos los cuales juegan un papel en el riesgo individual de toxicidad y cáncer.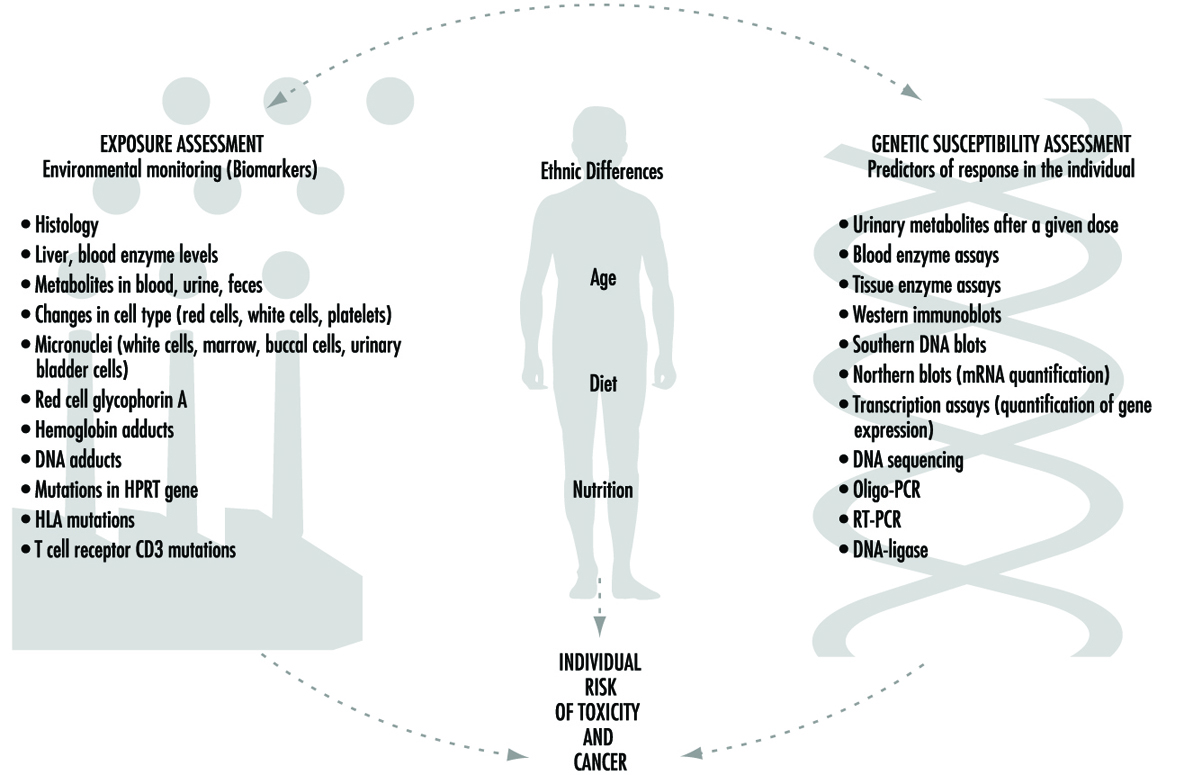
Primero describiremos la variabilidad de las respuestas tóxicas en humanos. Luego presentaremos algunas de las enzimas responsables de tal variación en la respuesta, debido a diferencias en el metabolismo de sustancias químicas extrañas. A continuación, se detallará la historia y nomenclatura de la superfamilia del citocromo P450. Se describirán brevemente cinco polimorfismos P450 humanos así como varios polimorfismos no P450; estos son responsables de las diferencias humanas en la respuesta tóxica. Luego discutiremos un ejemplo para enfatizar el punto de que las diferencias genéticas en los individuos pueden influir en la evaluación de la exposición, según lo determinado por el monitoreo ambiental. Por último, discutiremos el papel de estas enzimas metabolizadoras de xenobióticos en funciones vitales críticas.
Variación en la respuesta tóxica entre la población humana
Toxicólogos y farmacólogos hablan comúnmente de la dosis letal promedio para el 50% de la población (LD50), la dosis máxima media tolerada por el 50% de la población (MTD50), y la dosis efectiva promedio de un fármaco en particular para el 50% de la población (ED50). Sin embargo, ¿cómo nos afectan estas dosis a cada uno de nosotros de forma individual? En otras palabras, un individuo altamente sensible puede verse 500 veces más afectado o 500 veces más propenso a verse afectado que el individuo más resistente de una población; para estas personas, el LD50 (y MTD50 y DE50) los valores tendrían poco significado. LD50, MTD50 y DE50 los valores sólo son relevantes cuando se refieren a la población en su conjunto.
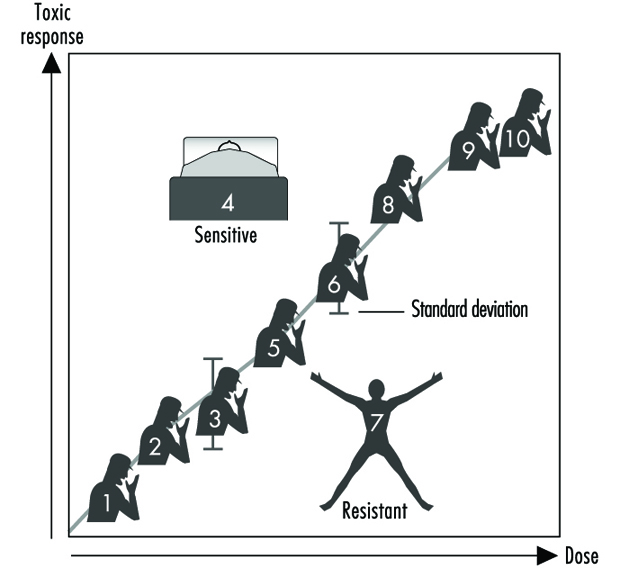
Figura 2 y XNUMX ilustra una relación dosis-respuesta hipotética para una respuesta tóxica de individuos en cualquier población dada. Este diagrama genérico podría representar el carcinoma broncogénico en respuesta a la cantidad de cigarrillos fumados, el cloracné en función de los niveles de dioxina en el lugar de trabajo, el asma en función de las concentraciones de ozono o aldehído en el aire, las quemaduras solares en respuesta a la luz ultravioleta, la disminución del tiempo de coagulación como una función de la ingesta de aspirina, o molestias gastrointestinales en respuesta al número de Jalapeño pimientos consumidos. Generalmente, en cada uno de estos casos, cuanto mayor sea la exposición, mayor será la respuesta tóxica. La mayor parte de la población exhibirá la media y la desviación estándar de la respuesta tóxica en función de la dosis. El "valor atípico resistente" (abajo a la derecha en la figura 2) es un individuo que tiene menos respuesta a dosis o exposiciones más altas. Un “valor atípico sensible” (arriba a la izquierda) es un individuo que tiene una respuesta exagerada a una dosis o exposición relativamente pequeña. Estos valores atípicos, con diferencias extremas en la respuesta en comparación con la mayoría de los individuos de la población, pueden representar variantes genéticas importantes que pueden ayudar a los científicos a intentar comprender los mecanismos moleculares subyacentes de una respuesta tóxica.
Figura 2. Relación genérica entre cualquier respuesta tóxica y la dosis de cualquier agente ambiental, químico o físico
Utilizando estos valores atípicos en estudios familiares, los científicos de varios laboratorios han comenzado a apreciar la importancia de la herencia mendeliana para una determinada respuesta tóxica. Posteriormente, se puede recurrir a la biología molecular y los estudios genéticos para identificar el mecanismo subyacente a nivel genético (genotipo) responsable de la enfermedad causada por el medio ambiente (fenotipo).
Enzimas metabolizadoras de xenobióticos o fármacos
¿Cómo responde el cuerpo a la miríada de sustancias químicas exógenas a las que estamos expuestos? Los humanos y otros mamíferos han desarrollado sistemas enzimáticos metabólicos altamente complejos que comprenden más de una docena de superfamilias distintas de enzimas. Casi todas las sustancias químicas a las que los humanos están expuestos serán modificadas por estas enzimas para facilitar la eliminación de la sustancia extraña del cuerpo. En conjunto, estas enzimas se denominan con frecuencia como enzimas metabolizadoras de fármacos or enzimas metabolizadoras de xenobióticos. En realidad, ambos términos son nombres inapropiados. En primer lugar, muchas de estas enzimas no solo metabolizan medicamentos, sino también cientos de miles de sustancias químicas dietéticas y ambientales. En segundo lugar, todas estas enzimas también tienen compuestos corporales normales como sustratos; ninguna de estas enzimas metaboliza solo sustancias químicas extrañas.
Durante más de cuatro décadas, los procesos metabólicos mediados por estas enzimas se han clasificado comúnmente como reacciones de Fase I o Fase II (figura 3). Las reacciones de fase I ("funcionalización") generalmente involucran modificaciones estructurales relativamente menores del químico original a través de oxidación, reducción o hidrólisis para producir un metabolito más soluble en agua. Con frecuencia, las reacciones de la Fase I proporcionan un "control" para la modificación adicional de un compuesto mediante reacciones posteriores de la Fase II. Las reacciones de fase I están mediadas principalmente por una superfamilia de enzimas muy versátiles, denominadas colectivamente citocromos P450, aunque también pueden participar otras superfamilias de enzimas (figura 4).
Figura 3. La designación clásica de las enzimas metabolizadoras de fármacos o xenobióticos de Fase I y Fase II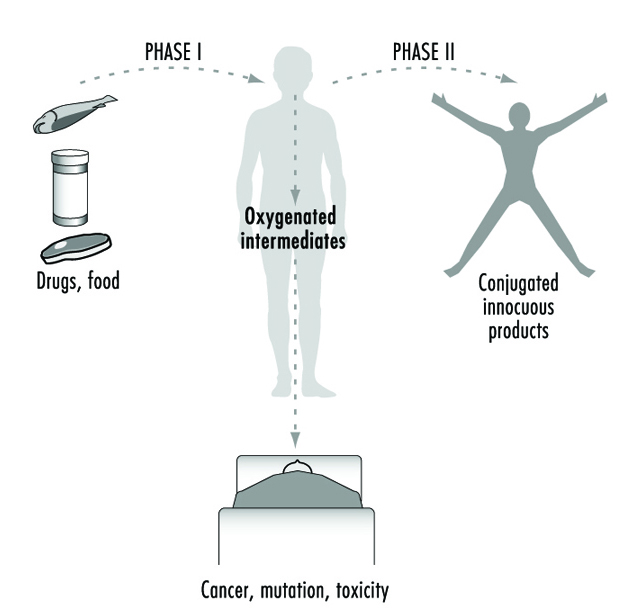
Figura 4. Ejemplos de enzimas metabolizadoras de fármacos
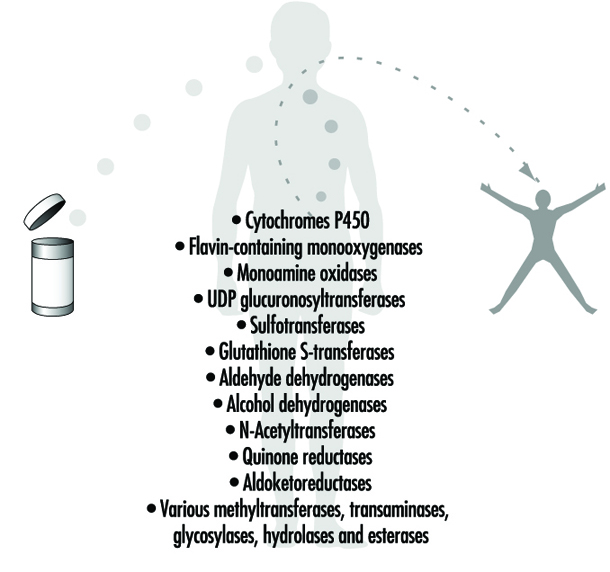
Las reacciones de fase II implican el acoplamiento de una molécula endógena soluble en agua a una sustancia química (sustancia química principal o metabolito de fase I) para facilitar la excreción. Las reacciones de fase II se denominan con frecuencia reacciones de "conjugación" o "derivatización". Las superfamilias de enzimas que catalizan las reacciones de la Fase II generalmente se nombran de acuerdo con el resto de conjugación endógeno involucrado: por ejemplo, acetilación por las N-acetiltransferasas, sulfatación por las sulfotransferasas, conjugación de glutatión por las glutatión transferasas y glucuronidación por las UDP glucuronosiltransferasas (figura 4) . Aunque el órgano principal del metabolismo de los fármacos es el hígado, los niveles de algunas enzimas metabolizadoras de fármacos son bastante altos en el tracto gastrointestinal, las gónadas, los pulmones, el cerebro y los riñones, y tales enzimas están indudablemente presentes en cierta medida en todas las células vivas.
Las enzimas metabolizadoras de xenobióticos representan un doble filo Swords
A medida que aprendemos más sobre los procesos biológicos y químicos que conducen a las aberraciones en la salud humana, se hace cada vez más evidente que las enzimas metabolizadoras de fármacos funcionan de manera ambivalente (figura 3). En la mayoría de los casos, las sustancias químicas solubles en lípidos se convierten en metabolitos solubles en agua que se excretan más fácilmente. Sin embargo, está claro que en muchas ocasiones las mismas enzimas son capaces de transformar otros químicos inertes en moléculas altamente reactivas. Estos intermediarios pueden luego interactuar con macromoléculas celulares como proteínas y ADN. Por lo tanto, para cada sustancia química a la que los seres humanos están expuestos, existe el potencial para las vías competitivas de activación metabólica y desintoxicación.
Breve repaso de la genética
En genética humana, cada gen (loci) se encuentra en uno de los 23 pares de cromosomas. Los dos alelos (uno presente en cada cromosoma del par) pueden ser iguales o pueden ser diferentes entre sí. por ejemplo, el B y b alelos, en los que B (ojos marrones) es dominante sobre b (ojos azules): los individuos del fenotipo de ojos marrones pueden tener el BB or Bb genotipos, mientras que los individuos del fenotipo de ojos azules solo pueden tener bb genotipo.
A polimorfismo se define como dos o más fenotipos (rasgos) heredados de manera estable, derivados de los mismos genes, que se mantienen en la población, a menudo por razones que no son necesariamente obvias. Para que un gen sea polimórfico, el producto del gen no debe ser esencial para el desarrollo, el vigor reproductivo u otros procesos vitales críticos. De hecho, un "polimorfismo equilibrado", en el que el heterocigoto tiene una clara ventaja de supervivencia sobre cualquiera de los homocigotos (p. ej., resistencia a la malaria y al alelo de la hemoglobina de células falciformes) es una explicación común para mantener un alelo en la población en niveles altos que de otro modo no se explicarían. frecuencias (ver González y Nebert 1990).
Polimorfismos humanos de enzimas metabolizadoras de xenobióticos
Las diferencias genéticas en el metabolismo de varios fármacos y sustancias químicas ambientales se conocen desde hace más de cuatro décadas (Kalow 1962 y 1992). Estas diferencias se denominan con frecuencia como farmacogenético o, más ampliamente, polimorfismos ecogenéticos. Estos polimorfismos representan alelos variantes que ocurren con una frecuencia relativamente alta en la población y generalmente están asociados con aberraciones en la expresión o función de la enzima. Históricamente, los polimorfismos generalmente se identificaban después de respuestas inesperadas a agentes terapéuticos. Más recientemente, la tecnología del ADN recombinante ha permitido a los científicos identificar las alteraciones precisas en los genes que son responsables de algunos de estos polimorfismos. Los polimorfismos ahora se han caracterizado en muchas enzimas metabolizadoras de fármacos, incluidas las enzimas de fase I y fase II. A medida que se identifican más y más polimorfismos, se hace cada vez más evidente que cada individuo puede poseer un complemento distinto de enzimas metabolizadoras de fármacos. Esta diversidad podría describirse como una "huella digital metabólica". Es la interacción compleja de las diversas superfamilias de enzimas metabolizadoras de drogas dentro de cualquier individuo lo que finalmente determinará su respuesta particular a un químico dado (Kalow 1962 y 1992; Nebert 1988; Gonzalez y Nebert 1990; Nebert y Weber 1990).
Expresión de enzimas metabolizadoras de xenobióticos humanos en células Cultura
¿Cómo podríamos desarrollar mejores predictores de las respuestas tóxicas humanas a los productos químicos? Los avances en la definición de la multiplicidad de enzimas que metabolizan fármacos deben ir acompañados de un conocimiento preciso de qué enzimas determinan el destino metabólico de sustancias químicas individuales. Los datos recopilados de los estudios de laboratorio con roedores sin duda han proporcionado información útil. Sin embargo, las diferencias significativas entre especies en las enzimas metabolizadoras de xenobióticos requieren precaución al extrapolar los datos a las poblaciones humanas. Para superar esta dificultad, muchos laboratorios han desarrollado sistemas en los que se pueden diseñar varias líneas celulares en cultivo para producir enzimas humanas funcionales que son estables y en altas concentraciones (González, Crespi y Gelboin 1991). La producción exitosa de enzimas humanas se ha logrado en una variedad de diversas líneas celulares de fuentes que incluyen bacterias, levaduras, insectos y mamíferos.
Para definir el metabolismo de los productos químicos con mayor precisión, múltiples enzimas también se han producido con éxito en una sola línea celular (González, Crespi y Gelboin 1991). Estas líneas celulares brindan información valiosa sobre las enzimas precisas involucradas en el procesamiento metabólico de cualquier compuesto dado y metabolitos tóxicos probables. Si esta información se puede combinar con el conocimiento sobre la presencia y el nivel de una enzima en los tejidos humanos, estos datos deberían proporcionar valiosos predictores de respuesta.
Cytochrome P450
Historia y nomenclatura
La superfamilia del citocromo P450 es una de las superfamilias de enzimas metabolizadoras de fármacos más estudiadas y tiene una gran variabilidad individual en respuesta a sustancias químicas. Citocromo P450 es un término genérico conveniente que se usa para describir una gran superfamilia de enzimas fundamentales en el metabolismo de innumerables sustratos endógenos y exógenos. El termino citocromo P450 fue acuñado por primera vez en 1962 para describir un desconocido pigmentación en células que, cuando se redujeron y se unieron con monóxido de carbono, produjeron un pico de absorción característico a 450 nm. Desde principios de la década de 1980, la tecnología de clonación de cDNA ha dado lugar a importantes conocimientos sobre la multiplicidad de enzimas del citocromo P450. Hasta la fecha, se han identificado más de 400 genes distintos del citocromo P450 en animales, plantas, bacterias y levaduras. Se ha estimado que cualquier especie de mamífero, como los humanos, puede poseer 60 o más genes P450 distintos (Nebert y Nelson 1991). La multiplicidad de genes P450 ha requerido el desarrollo de un sistema de nomenclatura estandarizado (Nebert et al. 1987; Nelson et al. 1993). Propuesto por primera vez en 1987 y actualizado cada dos años, el sistema de nomenclatura se basa en la evolución divergente de las comparaciones de secuencias de aminoácidos entre las proteínas P450. Los genes P450 se dividen en familias y subfamilias: las enzimas dentro de una familia muestran una similitud de aminoácidos superior al 40 %, y las de la misma subfamilia muestran una similitud del 55 %. Los genes P450 se nombran con el símbolo de raíz CYP seguido de un número arábigo que designa la familia P450, una letra que indica la subfamilia y otro número arábigo que designa el gen individual (Nelson et al. 1993; Nebert et al. 1991). Por lo tanto, CYP1A1 representa el gen 450 de P1 en la familia 1 y la subfamilia A.
Hasta febrero de 1995, hay 403 CYP genes en la base de datos, compuesta por 59 familias y 105 subfamilias. Estos incluyen ocho familias de eucariotas inferiores, 15 familias de plantas y 19 familias de bacterias. Las 15 familias de genes P450 humanos comprenden 26 subfamilias, 22 de las cuales se han mapeado en ubicaciones cromosómicas en la mayor parte del genoma. Algunas secuencias son claramente ortólogas en muchas especies, por ejemplo, solo una CYP17 (esteroide 17α-hidroxilasa) se ha encontrado en todos los vertebrados examinados hasta la fecha; otras secuencias dentro de una subfamilia están muy duplicadas, lo que hace imposible la identificación de pares ortólogos (p. ej., el CYP2C subfamilia). Curiosamente, el ser humano y la levadura comparten un gen ortólogo en el CYP51 familia. Numerosas revisiones exhaustivas están disponibles para los lectores que buscan más información sobre la superfamilia P450 (Nelson et al. 1993; Nebert et al. 1991; Nebert y McKinnon 1994; Guengerich 1993; Gonzalez 1992).
El éxito del sistema de nomenclatura P450 ha resultado en el desarrollo de sistemas terminológicos similares para las glucuronosiltransferasas UDP (Burchell et al. 1991) y las monooxigenasas que contienen flavina (Lawton et al. 1994). También se están desarrollando sistemas de nomenclatura similares basados en la evolución divergente para otras superfamilias de enzimas metabolizadoras de fármacos (p. ej., sulfotransferasas, epóxido hidrolasas y aldehído deshidrogenasas).
Recientemente, dividimos la superfamilia de genes P450 de mamíferos en tres grupos (Nebert y McKinnon 1994): los involucrados principalmente en el metabolismo químico extraño, los involucrados en la síntesis de varias hormonas esteroides y los que participan en otras funciones endógenas importantes. Son las enzimas P450 metabolizadoras de xenobióticos las que asumen la mayor importancia para la predicción de la toxicidad.
Enzimas P450 que metabolizan xenobióticos
Las enzimas P450 involucradas en el metabolismo de compuestos extraños y fármacos casi siempre se encuentran dentro de las familias. CYP1, CYP2, CYP3 y CYP4. Estas enzimas P450 catalizan una amplia variedad de reacciones metabólicas, con un solo P450 a menudo capaz de metabolizar muchos compuestos diferentes. Además, múltiples enzimas P450 pueden metabolizar un solo compuesto en diferentes sitios. Además, un compuesto puede ser metabolizado en el mismo sitio único por varios P450, aunque a velocidades variables.
Una propiedad muy importante de las enzimas P450 que metabolizan fármacos es que muchos de estos genes son inducibles por las mismas sustancias que sirven como sus sustratos. Por otro lado, otros genes P450 son inducidos por no sustratos. Este fenómeno de inducción enzimática es la base de muchas interacciones farmacológicas de importancia terapéutica.
Aunque están presentes en muchos tejidos, estas enzimas P450 particulares se encuentran en niveles relativamente altos en el hígado, el sitio principal del metabolismo de los fármacos. Algunas de las enzimas P450 que metabolizan xenobióticos exhiben actividad hacia ciertos sustratos endógenos (p. ej., ácido araquidónico). Sin embargo, en general se cree que la mayoría de estas enzimas P450 que metabolizan xenobióticos no desempeñan funciones fisiológicas importantes, aunque esto aún no se ha establecido experimentalmente. Es probable que la disrupción homocigota selectiva, o “knock-out”, de genes P450 metabolizadores de xenobióticos individuales por medio de metodologías dirigidas a genes en ratones probablemente proporcione pronto información inequívoca con respecto a las funciones fisiológicas de los P450 metabolizadores de xenobióticos (para una revisión de selección de genes, véase Capecchi 1994).
En contraste con las familias P450 que codifican enzimas involucradas principalmente en procesos fisiológicos, las familias que codifican enzimas P450 que metabolizan xenobióticos muestran una marcada especificidad de especie y frecuentemente contienen muchos genes activos por subfamilia (Nelson et al. 1993; Nebert et al. 1991). Dada la aparente falta de sustratos fisiológicos, es posible que las enzimas P450 en las familias CYP1, CYP2, CYP3 y CYP4 que han aparecido en los últimos cientos de millones de años han evolucionado como un medio de desintoxicación de sustancias químicas extrañas que se encuentran en el medio ambiente y la dieta. Claramente, la evolución de los P450 que metabolizan xenobióticos habría ocurrido durante un período de tiempo que precede con mucho a la síntesis de la mayoría de los productos químicos sintéticos a los que los humanos están expuestos ahora. Los genes en estas cuatro familias de genes pueden haber evolucionado y divergido en los animales debido a su exposición a los metabolitos de las plantas durante los últimos 1.2 millones de años, un proceso denominado descriptivamente "guerra animal-planta" (González y Nebert 1990). La guerra animal-planta es el fenómeno en el que las plantas desarrollan nuevos químicos (fitoalexinas) como un mecanismo de defensa para evitar que los animales los ingieran, y los animales, a su vez, responden desarrollando nuevos genes P450 para adaptarse a los sustratos diversificados. Los ejemplos recientemente descritos de guerra química planta-insecto y planta-hongo que involucran la desintoxicación de sustratos tóxicos con P450 dan un mayor impulso a esta propuesta (Nebert 1994).
La siguiente es una breve introducción a varios de los polimorfismos de la enzima P450 que metaboliza xenobióticos humanos en los que se cree que los determinantes genéticos de la respuesta tóxica son de gran importancia. Hasta hace poco, los polimorfismos de P450 generalmente se sugerían por una variación inesperada en la respuesta del paciente a los agentes terapéuticos administrados. De hecho, varios polimorfismos P450 se nombran de acuerdo con el fármaco con el que se identificó por primera vez el polimorfismo. Más recientemente, los esfuerzos de investigación se han centrado en la identificación de las enzimas P450 precisas implicadas en el metabolismo de las sustancias químicas para las que se observa variación y la caracterización precisa de los genes P450 implicados. Como se describió anteriormente, la actividad medible de una enzima P450 hacia una sustancia química modelo puede denominarse fenotipo. Las diferencias alélicas en un gen P450 para cada individuo se denominan genotipo P450. A medida que se aplica más y más escrutinio al análisis de los genes P450, la base molecular precisa de la variación fenotípica previamente documentada se vuelve más clara.
La subfamilia CYP1A
El CYP1A La subfamilia comprende dos enzimas en humanos y todos los demás mamíferos: estos se designan CYP1A1 y CYP1A2 bajo la nomenclatura estándar P450. Estas enzimas son de gran interés porque están involucradas en la activación metabólica de muchos procarcinógenos y también son inducidas por varios compuestos de interés toxicológico, incluida la dioxina. Por ejemplo, CYP1A1 activa metabólicamente muchos compuestos que se encuentran en el humo del cigarrillo. CYP1A2 activa metabólicamente muchas arilaminas, asociadas con el cáncer de vejiga urinaria, que se encuentran en la industria de tintes químicos. CYP1A2 también activa metabólicamente la 4-(metilnitrosamino)-1-(3-piridil)-1-butanona (NNK), una nitrosamina derivada del tabaco. CYP1A1 y CYP1A2 también se encuentran en niveles más altos en los pulmones de los fumadores de cigarrillos, debido a la inducción de los hidrocarburos policíclicos presentes en el humo. Por lo tanto, se considera que los niveles de actividad de CYP1A1 y CYP1A2 son determinantes importantes de la respuesta individual a muchas sustancias químicas potencialmente tóxicas.
Interés toxicológico en el CYP1A subfamilia se intensificó en gran medida por un informe de 1973 que correlacionó el nivel de inducibilidad de CYP1A1 en fumadores de cigarrillos con la susceptibilidad individual al cáncer de pulmón (Kellermann, Shaw y Luyten-Kellermann 1973). La base molecular de la inducción de CYP1A1 y CYP1A2 ha sido un foco importante de numerosos laboratorios. El proceso de inducción está mediado por una proteína denominada receptor Ah a la que se unen las dioxinas y las sustancias químicas estructuralmente relacionadas. El nombre Ah se deriva de la aryl hnaturaleza hidrocarbonada de muchos inductores de CYP1A. Curiosamente, las diferencias en el gen que codifica el receptor Ah entre cepas de ratones dan como resultado marcadas diferencias en la respuesta química y la toxicidad. Un polimorfismo en el gen del receptor Ah también parece ocurrir en humanos: aproximadamente una décima parte de la población muestra una alta inducción de CYP1A1 y puede tener un mayor riesgo que las otras nueve décimas partes de desarrollar ciertos cánceres inducidos químicamente. El papel del receptor Ah en el control de las enzimas en el CYP1A subfamilia, y su papel como determinante de la respuesta humana a la exposición química, ha sido objeto de varias revisiones recientes (Nebert, Petersen y Puga 1991; Nebert, Puga y Vasiliou 1993).
¿Existen otros polimorfismos que puedan controlar el nivel de proteínas CYP1A en una célula? Un polimorfismo en el CYP1A1 También se ha identificado un gen, y esto parece influir en el riesgo de cáncer de pulmón entre los fumadores de cigarrillos japoneses, aunque este mismo polimorfismo no parece influir en el riesgo en otros grupos étnicos (Nebert y McKinnon 1994).
CYP2C19
Las variaciones en la velocidad a la que los individuos metabolizan el fármaco anticonvulsivo (S)-mefenitoína han sido bien documentadas durante muchos años (Guengerich 1989). Entre el 2% y el 5% de los caucásicos y hasta el 25% de los asiáticos son deficientes en esta actividad y pueden tener un mayor riesgo de toxicidad por el fármaco. Hace tiempo que se sabe que este defecto enzimático involucra a un miembro del ser humano CYP2C subfamilia, pero la base molecular precisa de esta deficiencia ha sido objeto de considerable controversia. La razón principal de esta dificultad fueron los seis o más genes en el ser humano. CYP2C subfamilia. Sin embargo, recientemente se demostró que una mutación de una sola base en el CYP2C19 gen es la causa principal de esta deficiencia (Goldstein y de Morais 1994). También se ha desarrollado una prueba simple de ADN, basada en la reacción en cadena de la polimerasa (PCR), para identificar rápidamente esta mutación en poblaciones humanas (Goldstein y de Morais 1994).
CYP2D6
Quizás la variación caracterizada más extensamente en un gen P450 es la que involucra al CYP2D6 gene. Se han descrito más de una docena de ejemplos de mutaciones, reordenamientos y deleciones que afectan a este gen (Meyer 1994). Este polimorfismo fue sugerido por primera vez hace 20 años por la variabilidad clínica en la respuesta de los pacientes al agente antihipertensivo detritioquina. Alteraciones en el CYP2D6 gen que da lugar a una actividad enzimática alterada se denominan colectivamente polimorfismo de la ruinoquina.
Antes de la llegada de los estudios basados en el ADN, las personas se clasificaban como metabolizadores lentos o rápidos (PM, EM) de la ruinoquina en función de las concentraciones de metabolitos en las muestras de orina. Ahora está claro que las alteraciones en el CYP2D6 El gen puede dar como resultado que los individuos muestren no solo un metabolismo pobre o extenso de la ruinoquina, sino también un metabolismo ultrarrápido. La mayoría de las alteraciones en el CYP2D6 están asociados con deficiencia parcial o total de la función enzimática; sin embargo, recientemente se han descrito individuos en dos familias que poseen múltiples copias funcionales del CYP2D6 gen, dando lugar al metabolismo ultrarrápido de los sustratos de CYP2D6 (Meyer 1994). Esta notable observación proporciona nuevos conocimientos sobre el amplio espectro de actividad de CYP2D6 observado previamente en estudios de población. Las alteraciones en la función de CYP2D6 son de particular importancia, dado que esta enzima metaboliza más de 30 fármacos comúnmente prescritos. Por lo tanto, la función CYP2D6 de un individuo es un factor determinante de la respuesta terapéutica y tóxica a la terapia administrada. De hecho, recientemente se ha argumentado que la consideración del estado de CYP2D6 de un paciente es necesaria para el uso seguro de medicamentos psiquiátricos y cardiovasculares.
El papel de la CYP2D6 El polimorfismo como determinante de la susceptibilidad individual a enfermedades humanas como el cáncer de pulmón y la enfermedad de Parkinson también ha sido objeto de intensos estudios (Nebert y McKinnon 1994; Meyer 1994). Si bien las conclusiones son difíciles de definir dada la naturaleza diversa de los protocolos de estudio utilizados, la mayoría de los estudios parecen indicar una asociación entre los metabolizadores rápidos de la ruinoquina (fenotipo EM) y el cáncer de pulmón. Las razones de tal asociación no están claras actualmente. Sin embargo, se ha demostrado que la enzima CYP2D6 metaboliza la NNK, una nitrosamina derivada del tabaco.
A medida que mejoren los ensayos basados en ADN, lo que permite una evaluación aún más precisa del estado de CYP2D6, se anticipa que se aclarará la relación precisa de CYP2D6 con el riesgo de enfermedad. Mientras que el metabolizador rápido puede estar relacionado con la susceptibilidad al cáncer de pulmón, el metabolizador lento (fenotipo PM) parece estar asociado con la enfermedad de Parkinson de causa desconocida. Si bien estos estudios también son difíciles de comparar, parece que los individuos con PM que tienen una capacidad disminuida para metabolizar los sustratos de CYP2D6 (por ejemplo, la ruinoquina) tienen un riesgo de 2 a 2.5 veces mayor de desarrollar la enfermedad de Parkinson.
CYP2E1
El CYP2E1 El gen codifica una enzima que metaboliza muchas sustancias químicas, incluidos fármacos y muchos carcinógenos de bajo peso molecular. Esta enzima también es de interés porque es altamente inducible por el alcohol y puede desempeñar un papel en la lesión hepática inducida por sustancias químicas como el cloroformo, el cloruro de vinilo y el tetracloruro de carbono. La enzima se encuentra principalmente en el hígado y el nivel de enzima varía notablemente entre los individuos. Escrutinio minucioso de la CYP2E1 gen ha resultado en la identificación de varios polimorfismos (Nebert y McKinnon 1994). Se ha reportado una relación entre la presencia de ciertas variaciones estructurales en la CYP2E1 gen y riesgo aparentemente reducido de cáncer de pulmón en algunos estudios; sin embargo, existen claras diferencias interétnicas que requieren aclarar esta posible relación.
La subfamilia CYP3A
En humanos, se han identificado cuatro enzimas como miembros de la CYP3A subfamilia debido a su similitud en la secuencia de aminoácidos. Las enzimas CYP3A metabolizan muchos medicamentos comúnmente recetados, como la eritromicina y la ciclosporina. El contaminante cancerígeno alimentario aflatoxina B1 también es un sustrato de CYP3A. Un miembro del ser humano CYP3A subfamilia, designada CYP3A4, es el principal P450 en el hígado humano además de estar presente en el tracto gastrointestinal. Como ocurre con muchas otras enzimas P450, el nivel de CYP3A4 es muy variable entre los individuos. Una segunda enzima, denominada CYP3A5, se encuentra en solo aproximadamente el 25% de los hígados; la base genética de este hallazgo no ha sido dilucidada. Aún no se ha establecido la importancia de la variabilidad de CYP3A4 o CYP3A5 como factor en los determinantes genéticos de la respuesta tóxica (Nebert y McKinnon 1994).
Polimorfismos no P450
También existen numerosos polimorfismos dentro de otras superfamilias de enzimas metabolizadoras de xenobióticos (p. ej., glutatión transferasas, UDP glucuronosiltransferasas, paraoxonasas, deshidrogenasas, N-acetiltransferasas y monooxigenasas que contienen flavina). Debido a que la toxicidad final de cualquier intermedio generado por P450 depende de la eficiencia de las reacciones de desintoxicación de fase II posteriores, el papel combinado de múltiples polimorfismos enzimáticos es importante para determinar la susceptibilidad a enfermedades inducidas químicamente. Por lo tanto, es probable que el equilibrio metabólico entre las reacciones de fase I y fase II (figura 3) sea un factor importante en las enfermedades humanas inducidas químicamente y los determinantes genéticos de la respuesta tóxica.
El polimorfismo del gen GSTM1
Un ejemplo bien estudiado de un polimorfismo en una enzima de Fase II es el que involucra a un miembro de la superfamilia de enzimas glutatión S-transferasa, denominada GST mu o GSTM1. Esta enzima en particular tiene un interés toxicológico considerable porque parece estar involucrada en la desintoxicación subsiguiente de metabolitos tóxicos producidos a partir de sustancias químicas en el humo del cigarrillo por la enzima CYP1A1. El polimorfismo identificado en este gen de la glutatión transferasa implica una ausencia total de enzima funcional en hasta la mitad de todos los caucásicos estudiados. Esta falta de una enzima de Fase II parece estar asociada con una mayor susceptibilidad al cáncer de pulmón. Al agrupar a los individuos sobre la base de ambas variantes CYP1A1 genes y la deleción o presencia de un funcional GSTM1 gen, se ha demostrado que el riesgo de desarrollar cáncer de pulmón inducido por fumar varía significativamente (Kawajiri, Watanabe y Hayashi 1994). En particular, las personas que muestran una rara CYP1A1 alteración del gen, en combinación con la ausencia del GSTM1 tenían un riesgo mayor (hasta nueve veces) de desarrollar cáncer de pulmón cuando se exponían a un nivel relativamente bajo de humo de cigarrillo. Curiosamente, parece haber diferencias interétnicas en la importancia de los genes variantes que requieren más estudios para dilucidar el papel preciso de tales alteraciones en la susceptibilidad a la enfermedad (Kalow 1962; Nebert y McKinnon 1994; Kawajiri, Watanabe y Hayashi 1994).
Efecto sinérgico de dos o más polimorfismos sobre el tóxico. respuesta
Una respuesta tóxica a un agente ambiental puede ser muy exagerada por la combinación de dos defectos farmacogenéticos en el mismo individuo, por ejemplo, los efectos combinados del polimorfismo N-acetiltransferasa (NAT2) y el polimorfismo glucosa-6-fosfato deshidrogenasa (G6PD) .
La exposición ocupacional a las arilaminas constituye un grave riesgo de cáncer de vejiga urinaria. Desde los elegantes estudios de Cartwright en 1954, ha quedado claro que el estado del N-acetilador es un factor determinante del cáncer de vejiga inducido por colorantes azoicos. Existe una correlación muy significativa entre el fenotipo acetilador lento y la aparición de cáncer de vejiga, así como el grado de invasividad de este cáncer en la pared de la vejiga. Por el contrario, existe una asociación significativa entre el fenotipo acetilador rápido y la incidencia de carcinoma colorrectal. La N-acetiltransferasa (NAT1, NAT2) se han clonado y secuenciado, y los ensayos basados en ADN ahora pueden detectar más de una docena de variantes alélicas que explican el fenotipo de acetilador lento. Él NAT2 El gen es polimórfico y responsable de la mayor parte de la variabilidad en la respuesta tóxica a los químicos ambientales (Weber 1987; Grant 1993).
La glucosa-6-fosfato deshidrogenasa (G6PD) es una enzima crítica en la generación y mantenimiento de NADPH. La actividad baja o nula de G6PD puede conducir a una hemólisis grave inducida por fármacos o xenobióticos, debido a la ausencia de niveles normales de glutatión reducido (GSH) en los glóbulos rojos. La deficiencia de G6PD afecta al menos a 300 millones de personas en todo el mundo. Más del 10% de los hombres afroamericanos exhiben el fenotipo menos severo, mientras que ciertas comunidades sardas exhiben el “tipo mediterráneo” más severo con frecuencias tan altas como una de cada tres personas. Él G6PD El gen ha sido clonado y localizado en el cromosoma X, y numerosas mutaciones puntuales diversas explican el alto grado de heterogeneidad fenotípica observada en individuos con deficiencia de G6PD (Beutler 1992).
Se descubrió que la tiozalsulfona, un fármaco arilamina sulfa, provoca una distribución bimodal de la anemia hemolítica en la población tratada. Cuando se tratan con ciertos medicamentos, los individuos con la combinación de deficiencia de G6PD más el fenotipo de acetilador lento se ven más afectados que aquellos con la deficiencia de G6PD sola o el fenotipo de acetilador lento solo. Los acetiladores lentos con deficiencia de G6PD son al menos 40 veces más susceptibles que los acetiladores rápidos con G6PD normal a la hemólisis inducida por tiozalsulfona.
Efecto de los polimorfismos genéticos en la evaluación de la exposición
La evaluación de la exposición y el biomonitoreo (figura 1) también requieren información sobre la composición genética de cada individuo. Dada una exposición idéntica a un químico peligroso, el nivel de aductos de hemoglobina (u otros biomarcadores) puede variar en dos o tres órdenes de magnitud entre individuos, dependiendo de la huella digital metabólica de cada persona.
La misma farmacogenética combinada se ha estudiado en trabajadores de fábricas químicas en Alemania (tabla 1). Los aductos de hemoglobina entre los trabajadores expuestos a la anilina y la acetanilida son, con mucho, los más altos en los acetiladores lentos con deficiencia de G6PD, en comparación con los otros posibles fenotipos farmacogenéticos combinados. Este estudio tiene implicaciones importantes para la evaluación de la exposición. Estos datos demuestran que, aunque dos personas pueden estar expuestas al mismo nivel ambiental de sustancias químicas peligrosas en el lugar de trabajo, la cantidad de exposición (a través de biomarcadores como los aductos de hemoglobina) podría estimarse en dos o más órdenes de magnitud menos, debido a la predisposición genética subyacente del individuo. Asimismo, el riesgo resultante de un efecto adverso para la salud puede variar en dos o más órdenes de magnitud.
Tabla 1: Aductos de hemoglobina en trabajadores expuestos a anilina y acetanilida
| Estado del acetilador | Deficiencia de G6PD | |||
| Rápido | Lenta | No | Sí | Aductos de Hgb |
| + | + | 2 | ||
| + | + | 30 | ||
| + | + | 20 | ||
| + | + | 100 | ||
Fuente: Adaptado de Lewalter y Korallus 1985.
Diferencias genéticas en la unión y el metabolismo.
Debe enfatizarse que el mismo caso hecho aquí para el metabolismo también se puede hacer para la unión. Las diferencias hereditarias en la unión de los agentes ambientales afectarán en gran medida la respuesta tóxica. Por ejemplo, las diferencias en el mouse cdm El gen puede afectar profundamente la sensibilidad individual a la necrosis testicular inducida por cadmio (Taylor, Heiniger y Meier 1973). Es probable que las diferencias en la afinidad de unión del receptor Ah afecten la toxicidad inducida por dioxinas y el cáncer (Nebert, Petersen y Puga 1991; Nebert, Puga y Vasiliou 1993).
La figura 5 resume el papel del metabolismo y la unión en la toxicidad y el cáncer. Los agentes tóxicos, tal como existen en el medio ambiente o después del metabolismo o la unión, provocan sus efectos por una vía genotóxica (en la que se produce daño en el ADN) o una vía no genotóxica (en la que no es necesario que se produzca daño en el ADN ni mutagénesis). Curiosamente, recientemente quedó claro que los agentes "clásicos" que dañan el ADN pueden operar a través de una vía de transducción de señales no genotóxicas dependiente de glutatión reducido (GSH), que se inicia en o cerca de la superficie celular en ausencia de ADN y fuera del núcleo celular. (Devary y col. 1993). Sin embargo, las diferencias genéticas en el metabolismo y la unión siguen siendo los principales determinantes en el control de las diferentes respuestas tóxicas individuales.
Figura 5. Los medios generales por los que se produce la toxicidad
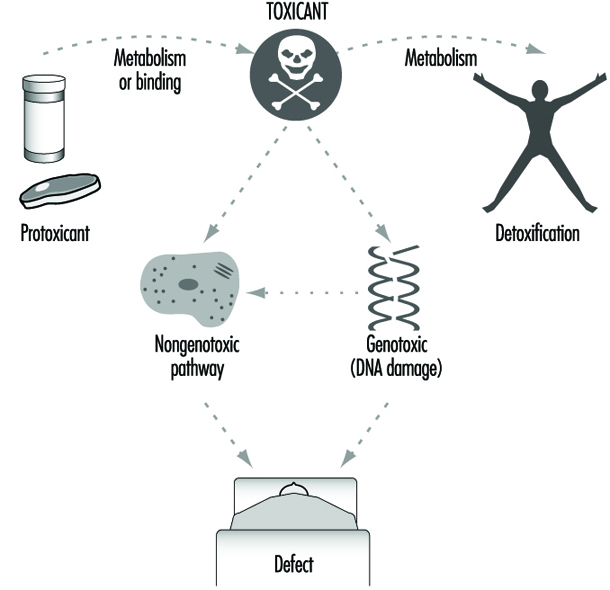
Papel de la función celular de enzimas metabolizadoras de fármacos
La variación de base genética en la función de las enzimas metabolizadoras de fármacos es de gran importancia para determinar la respuesta individual a las sustancias químicas. Estas enzimas son fundamentales para determinar el destino y el curso temporal de una sustancia química extraña después de la exposición.
Como se ilustra en la figura 5, la importancia de las enzimas metabolizadoras de fármacos en la susceptibilidad individual a la exposición química puede, de hecho, presentar un problema mucho más complejo de lo que es evidente a partir de esta simple discusión sobre el metabolismo de los xenobióticos. En otras palabras, durante las últimas dos décadas, los mecanismos genotóxicos (mediciones de aductos de ADN y aductos de proteínas) se han enfatizado mucho. Sin embargo, ¿qué pasa si los mecanismos no genotóxicos son al menos tan importantes como los mecanismos genotóxicos para causar respuestas tóxicas?
Como se mencionó anteriormente, las funciones fisiológicas de muchas enzimas metabolizadoras de fármacos involucradas en el metabolismo de xenobióticos no se han definido con precisión. Nebert (1994) ha propuesto que, debido a su presencia en este planeta durante más de 3.5 millones de años, las enzimas metabolizadoras de fármacos fueron originalmente (y ahora siguen siendo principalmente) responsables de regular los niveles celulares de muchos ligandos no peptídicos importantes en la activación transcripcional. de genes que afectan el crecimiento, la diferenciación, la apoptosis, la homeostasis y las funciones neuroendocrinas. Además, la toxicidad de la mayoría, si no de todos, los agentes ambientales se produce por medio de Agonista or antagonista acción sobre estas vías de transducción de señales (Nebert 1994). Con base en esta hipótesis, la variabilidad genética en las enzimas metabolizadoras de fármacos puede tener efectos bastante dramáticos en muchos procesos bioquímicos críticos dentro de la célula, lo que lleva a diferencias importantes en la respuesta tóxica. De hecho, es posible que tal escenario también pueda ser la base de muchas reacciones adversas idiosincrásicas encontradas en pacientes que usan medicamentos recetados comúnmente.
Conclusiones
La última década ha visto un progreso notable en nuestra comprensión de la base genética de la respuesta diferencial a los productos químicos en los medicamentos, los alimentos y los contaminantes ambientales. Las enzimas metabolizadoras de fármacos tienen una profunda influencia en la forma en que los seres humanos responden a las sustancias químicas. A medida que nuestra conciencia de la multiplicidad de enzimas metabolizadoras de fármacos sigue evolucionando, somos cada vez más capaces de realizar evaluaciones mejoradas del riesgo tóxico de muchos fármacos y productos químicos ambientales. Esto quizás se ilustra más claramente en el caso de la enzima citocromo P2 CYP6D450. Usando pruebas basadas en ADN relativamente simples, es posible predecir la respuesta probable de cualquier fármaco metabolizado predominantemente por esta enzima; esta predicción garantizará el uso más seguro de medicamentos valiosos, aunque potencialmente tóxicos.
El futuro sin duda verá una explosión en la identificación de más polimorfismos (fenotipos) que involucran enzimas metabolizadoras de fármacos. Esta información estará acompañada de pruebas mejoradas basadas en ADN mínimamente invasivas para identificar genotipos en poblaciones humanas.
Dichos estudios deberían ser particularmente informativos para evaluar el papel de los productos químicos en las muchas enfermedades ambientales de origen actualmente desconocido. La consideración de múltiples polimorfismos de enzimas metabolizadoras de fármacos, en combinación (p. ej., tabla 1), también es probable que represente un área de investigación particularmente fértil. Dichos estudios aclararán el papel de los productos químicos en la causalidad de los cánceres. Colectivamente, esta información debería permitir la formulación de consejos cada vez más individualizados sobre la evitación de productos químicos que probablemente sean de interés individual. Este es el campo de la toxicología preventiva. Sin duda, estos consejos ayudarán en gran medida a todas las personas a hacer frente a la carga química cada vez mayor a la que estamos expuestos.