Pratiquement toute la médecine est consacrée soit à prévenir la mort cellulaire, dans des maladies telles que l'infarctus du myocarde, les accidents vasculaires cérébraux, les traumatismes et les chocs, soit à la provoquer, comme dans le cas des maladies infectieuses et du cancer. Il est donc essentiel d'en comprendre la nature et les mécanismes impliqués. La mort cellulaire a été classée comme « accidentelle », c'est-à-dire causée par des agents toxiques, l'ischémie, etc., ou « programmée », comme cela se produit au cours du développement embryologique, y compris la formation des doigts et la résorption de la queue du têtard.
Les lésions cellulaires et la mort cellulaire sont donc importantes à la fois en physiologie et en physiopathologie. La mort cellulaire physiologique est extrêmement importante au cours de l'embryogenèse et du développement embryonnaire. L'étude de la mort cellulaire au cours du développement a conduit à des informations importantes et nouvelles sur la génétique moléculaire impliquée, notamment à travers l'étude du développement chez les animaux invertébrés. Chez ces animaux, la localisation précise et la signification des cellules destinées à subir la mort cellulaire ont été soigneusement étudiées et, grâce à l'utilisation des techniques classiques de mutagénèse, plusieurs gènes impliqués ont maintenant été identifiés. Dans les organes adultes, l'équilibre entre la mort cellulaire et la prolifération cellulaire contrôle la taille de l'organe. Dans certains organes, comme la peau et l'intestin, il y a un renouvellement continu des cellules. Dans la peau, par exemple, les cellules se différencient lorsqu'elles atteignent la surface, et subissent finalement une différenciation terminale et la mort cellulaire au fur et à mesure que la kératinisation se poursuit avec la formation d'enveloppes réticulées.
De nombreuses classes de produits chimiques toxiques sont capables d'induire des lésions cellulaires aiguës suivies de la mort. Ceux-ci comprennent l'anoxie et l'ischémie et leurs analogues chimiques tels que le cyanure de potassium ; les cancérigènes chimiques, qui forment des électrophiles qui se lient de manière covalente aux protéines des acides nucléiques ; des produits chimiques oxydants, entraînant la formation de radicaux libres et des lésions oxydantes ; activation du complément ; et une variété d'ionophores de calcium. La mort cellulaire est également une composante importante de la carcinogenèse chimique; de nombreux carcinogènes chimiques complets, à des doses cancérigènes, produisent une nécrose et une inflammation aiguës suivies d'une régénération et d'une prénéoplasie.
Définitions
Lésion cellulaire
Une lésion cellulaire est définie comme un événement ou un stimulus, tel qu'un produit chimique toxique, qui perturbe l'homéostasie normale de la cellule, provoquant ainsi un certain nombre d'événements (figure 1). Les principales cibles des lésions mortelles illustrées sont l'inhibition de la synthèse d'ATP, la perturbation de l'intégrité de la membrane plasmique ou le retrait des facteurs de croissance essentiels.
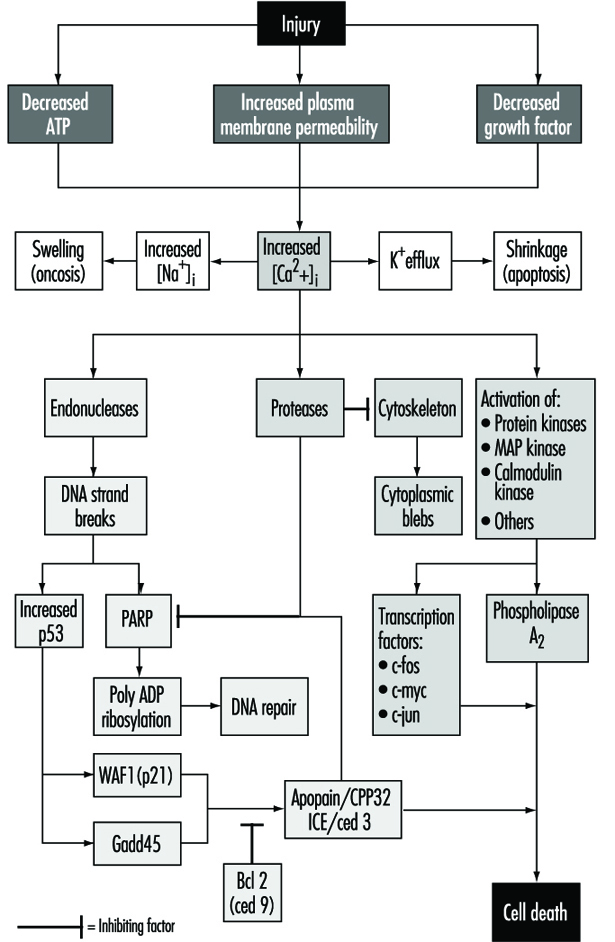
Les blessures mortelles entraînent la mort d'une cellule après une période de temps variable, en fonction de la température, du type de cellule et du stimulus ; ou ils peuvent être sublétaux ou chroniques, c'est-à-dire que la lésion entraîne une altération de l'homéostasie qui, bien qu'anormale, n'entraîne pas la mort cellulaire (Trump et Arstila 1971 ; Trump et Berezesky 1992 ; Trump et Berezesky 1995 ; Trump, Berezesky et Osornio-Vargas 1981). Dans le cas d'une blessure mortelle, il y a une phase avant le moment de la mort cellulaire
pendant ce temps, la cellule récupérera; cependant, après un moment donné (le "point de non-retour" ou le point de mort cellulaire), l'élimination de la blessure n'entraîne pas de récupération, mais la cellule subit une dégradation et une hydrolyse, atteignant finalement un équilibre physico-chimique avec le environnement. C'est la phase dite de nécrose. Au cours de la phase prélétale, plusieurs types principaux de changements se produisent, selon la cellule et le type de blessure. Celles-ci sont connues sous le nom d'apoptose et d'oncose.
L'apoptose
L'apoptose est dérivé des mots grecs apo, c'est-à-dire loin de, et ptosis, signifiant tomber. Le terme s'éloigner de vient du fait que, lors de ce type de changement prélétal, les cellules se rétractent et subissent un important bourgeonnement en périphérie. Les bulles se détachent alors et flottent. L'apoptose se produit dans une variété de types de cellules suite à divers types de lésions toxiques (Wyllie, Kerr et Currie 1980). Il est particulièrement important dans les lymphocytes, où il est le mécanisme prédominant de renouvellement des clones de lymphocytes. Les fragments résultants donnent les corps basophiles observés dans les macrophages des ganglions lymphatiques. Dans d'autres organes, l'apoptose se produit typiquement dans des cellules individuelles qui sont rapidement éliminées avant et après la mort par phagocytose des fragments par des cellules parenchymateuses adjacentes ou par des macrophages. L'apoptose survenant dans des cellules individuelles avec phagocytose ultérieure n'entraîne généralement pas d'inflammation. Avant la mort, les cellules apoptotiques présentent un cytosol très dense avec des mitochondries normales ou condensées. Le réticulum endoplasmique (RE) est normal ou peu dilaté. La chromatine nucléaire est nettement agglutinée le long de l'enveloppe nucléaire et autour du nucléole. Le contour nucléaire est également irrégulier et une fragmentation nucléaire se produit. La condensation de la chromatine est associée à la fragmentation de l'ADN qui, dans de nombreux cas, se produit entre les nucléosomes, donnant un aspect caractéristique en échelle lors de l'électrophorèse.
En apoptose, augmentation de [Ca2+]i peut stimuler K+ efflux entraînant un rétrécissement cellulaire, ce qui nécessite probablement de l'ATP. Les blessures qui inhibent totalement la synthèse d'ATP sont donc plus susceptibles d'entraîner l'apoptose. Une augmentation soutenue de [Ca2+]i a un certain nombre d'effets délétères, y compris l'activation des protéases, des endonucléases et des phospholipases. L'activation de l'endonucléase entraîne des ruptures de brins d'ADN simples et doubles qui, à leur tour, stimulent des niveaux accrus de p53 et de ribosylation poly-ADP, et de protéines nucléaires essentielles à la réparation de l'ADN. L'activation des protéases modifie un certain nombre de substrats, y compris l'actine et les protéines apparentées, conduisant à la formation de bulles. Un autre substrat important est la poly(ADP-ribose) polymérase (PARP), qui inhibe la réparation de l'ADN. Augmentation de [Ca2+]i est également associée à l'activation d'un certain nombre de protéines kinases, telles que la MAP kinase, la calmoduline kinase et autres. Ces kinases sont impliquées dans l'activation des facteurs de transcription qui initient la transcription des gènes précoces immédiats, par exemple, c-fos, c-jun et c-myc, et dans l'activation de la phospholipase A2 ce qui se traduit par une perméabilisation de la membrane plasmique et des membranes intracellulaires telles que la membrane interne des mitochondries.
Oncose
Oncose, dérivé du mot grec Est-ce que s, gonfler, est ainsi nommé parce que dans ce type de changement prélétal, la cellule commence à gonfler presque immédiatement après la blessure (Majno et Joris 1995). La raison du gonflement est une augmentation des cations dans l'eau à l'intérieur de la cellule. Le principal cation responsable est le sodium, qui est normalement régulé pour maintenir le volume cellulaire. Cependant, en l'absence d'ATP ou si la Na-ATPase du plasmalemme est inhibée, le contrôle du volume est perdu à cause des protéines intracellulaires et le sodium dans l'eau continue d'augmenter. Parmi les événements précoces de l'oncose sont donc augmentés [Na+]i ce qui conduit à un gonflement cellulaire et à une augmentation de [Ca2+]i résultant soit de l'influx de l'espace extracellulaire, soit de la libération des réserves intracellulaires. Il en résulte un gonflement du cytosol, un gonflement du réticulum endoplasmique et de l'appareil de Golgi, et la formation de bulles aqueuses autour de la surface cellulaire. Les mitochondries subissent initialement une condensation, mais plus tard, elles présentent également un gonflement de grande amplitude en raison de dommages à la membrane mitochondriale interne. Dans ce type de changement prélétal, la chromatine subit une condensation et finalement une dégradation ; cependant, le modèle d'échelle caractéristique de l'apoptose n'est pas observé.
Nécrose
La nécrose fait référence à la série de changements qui se produisent après la mort cellulaire lorsque la cellule est convertie en débris qui sont généralement éliminés par la réponse inflammatoire. Deux types peuvent être distingués : la nécrose oncotique et la nécrose apoptotique. La nécrose oncotique survient généralement dans de grandes zones, par exemple, dans un infarctus du myocarde ou régionalement dans un organe après une toxicité chimique, comme le tubule rénal proximal après administration de HgCl2. De larges zones d'un organe sont atteintes et les cellules nécrotiques provoquent rapidement une réaction inflammatoire, d'abord aiguë puis chronique. En cas de survie de l'organisme, dans de nombreux organes, la nécrose est suivie d'une élimination des cellules mortes et d'une régénération, par exemple dans le foie ou les reins suite à une toxicité chimique. En revanche, la nécrose apoptotique se produit généralement sur une seule cellule et les débris nécrotiques se forment dans les phagocytes des macrophages ou des cellules parenchymateuses adjacentes. Les premières caractéristiques des cellules nécrotiques comprennent des interruptions dans la continuité de la membrane plasmique et l'apparition de densités floconneuses, représentant des protéines dénaturées au sein de la matrice mitochondriale. Dans certaines formes de lésions qui n'interfèrent pas initialement avec l'accumulation de calcium mitochondrial, des dépôts de phosphate de calcium peuvent être observés dans les mitochondries. D'autres systèmes membranaires se fragmentent de la même manière, tels que le RE, les lysosomes et l'appareil de Golgi. En fin de compte, la chromatine nucléaire subit une lyse, résultant de l'attaque par les hydrolases lysosomales. Après la mort cellulaire, les hydrolases lysosomales jouent un rôle important dans l'élimination des débris avec les cathepsines, les nucléolases et les lipases, car celles-ci ont un pH acide optimal et peuvent survivre au faible pH des cellules nécrotiques tandis que d'autres enzymes cellulaires sont dénaturées et inactivées.
Mécanismes
Stimulus initial
Dans le cas de lésions mortelles, les interactions initiales les plus courantes entraînant une lésion entraînant la mort cellulaire sont l'interférence avec le métabolisme énergétique, comme l'anoxie, l'ischémie ou les inhibiteurs de la respiration, et la glycolyse comme le cyanure de potassium, le monoxyde de carbone, l'iodo-acétate et bientôt. Comme mentionné ci-dessus, des doses élevées de composés qui inhibent le métabolisme énergétique entraînent généralement une oncose. L'autre type courant de lésion initiale entraînant une mort cellulaire aiguë est la modification de la fonction de la membrane plasmique (Trump et Arstila 1971 ; Trump, Berezesky et Osornio-Vargas 1981). Cela peut être soit des dommages directs et une perméabilisation, comme dans le cas d'un traumatisme ou de l'activation du complexe C5b-C9 du complément, des dommages mécaniques à la membrane cellulaire ou une inhibition du sodium-potassium (Na+-K+) pompe avec des glycosides tels que l'ouabaïne. Les ionophores calciques tels que l'ionomycine ou A23187, qui transportent rapidement [Ca2+] vers le bas du gradient dans la cellule, provoquent également des blessures mortelles aiguës. Dans certains cas, le schéma du changement prélétal est l'apoptose ; dans d'autres, c'est une oncose.
Voies de signalisation
Avec de nombreux types de lésions, la respiration mitochondriale et la phosphorylation oxydative sont rapidement affectées. Dans certaines cellules, cela stimule la glycolyse anaérobie, qui est capable de maintenir l'ATP, mais avec de nombreuses blessures, cela est inhibé. Le manque d'ATP entraîne une incapacité à dynamiser un certain nombre de processus homéostatiques importants, en particulier le contrôle de l'homéostasie des ions intracellulaires (Trump et Berezesky 1992 ; Trump, Berezesky et Osornio-Vargas 1981). Il en résulte une augmentation rapide de [Ca2+]i, et augmenté [Na+] et [Cl-] entraîne un gonflement des cellules. Augmentation de [Ca2+]i entraîner l'activation d'un certain nombre d'autres mécanismes de signalisation discutés ci-dessous, y compris une série de kinases, ce qui peut entraîner une augmentation immédiate de la transcription précoce des gènes. Augmentation de [Ca2+]i modifie également la fonction cytosquelettique, entraînant en partie la formation de bulles et l'activation des endonucléases, des protéases et des phospholipases. Ceux-ci semblent déclencher bon nombre des effets importants discutés ci-dessus, tels que les dommages à la membrane par l'activation de la protéase et de la lipase, la dégradation directe de l'ADN à partir de l'activation de l'endonucléase et l'activation de kinases telles que la MAP kinase et la calmoduline kinase, qui agissent comme facteurs de transcription.
Grâce à un travail approfondi sur le développement chez les invertébrés C. elegans ainsi que Drosophila, ainsi que des cellules humaines et animales, une série de gènes pro-mort ont été identifiés. Certains de ces gènes d'invertébrés se sont avérés avoir des homologues de mammifères. Par exemple, le gène ced-3, essentiel à la mort cellulaire programmée chez C. elegans, a une activité protéase et une forte homologie avec l'enzyme de conversion de l'interleukine de mammifère (ICE). Un gène étroitement apparenté appelé apopain ou prICE a récemment été identifié avec une homologie encore plus étroite (Nicholson et al. 1995). Dans Drosophila, le gène reaper semble être impliqué dans un signal qui conduit à la mort cellulaire programmée. D'autres gènes pro-mort comprennent la protéine membranaire Fas et l'important gène suppresseur de tumeur, p53, qui est largement conservé. p53 est induit au niveau protéique suite à des dommages à l'ADN et, lorsqu'il est phosphorylé, agit comme un facteur de transcription pour d'autres gènes tels que gadd45 et waf-1, qui sont impliqués dans la signalisation de la mort cellulaire. D'autres gènes précoces immédiats tels que c-fos, c-jun et c-myc semblent également être impliqués dans certains systèmes.
En même temps, il existe des gènes anti-mort qui semblent contrecarrer les gènes pro-mort. Le premier d'entre eux à être identifié était ced-9 de C. elegans, qui est homologue à bcl-2 chez l'homme. Ces gènes agissent d'une manière encore inconnue pour empêcher la destruction des cellules par des toxines génétiques ou chimiques. Certaines preuves récentes indiquent que bcl-2 peut agir comme un antioxydant. Actuellement, de nombreux efforts sont en cours pour développer une compréhension des gènes impliqués et pour développer des moyens d'activer ou d'inhiber ces gènes, selon la situation.